To compare data of different samples, a
ModifierSet can be used. To select the data
alongside the transcripts and their positions a
GRanges or a
GRangesList needs to be provided.
In case of a GRanges object, the parent column must match the
transcript names as defined by the out put of ranges(x), whereas in
case of a GRangesList the element names must match the transcript
names.
compare(x, name, pos = 1L, ...)
compareByCoord(x, coord, ...)
plotCompare(x, name, pos = 1L, normalize, ...)
plotCompareByCoord(x, coord, normalize, ...)
# S4 method for class 'ModifierSet'
compare(x, name, pos = 1L, normalize, ...)
# S4 method for class 'ModifierSet,GRanges'
compareByCoord(x, coord, normalize, ...)
# S4 method for class 'ModifierSet,GRangesList'
compareByCoord(x, coord, normalize, ...)
# S4 method for class 'ModifierSet'
plotCompare(x, name, pos = 1L, normalize, ...)
# S4 method for class 'ModifierSet,GRanges'
plotCompareByCoord(x, coord, normalize, ...)
# S4 method for class 'ModifierSet,GRangesList'
plotCompareByCoord(x, coord, normalize, ...)Arguments
- x
a
ModifierorModifierSetobject.- name
Only for
compare: the transcript name- pos
Only for
compare: pos for comparison- ...
optional parameters:
alias: a data.frame with two columns,tx_idandname, to convert transcipt ids to another identifiername: Limit results to one specific gene or transcriptsequenceData: TRUE or FALSE? Should the aggregate of sequenceData be used for the comparison instead of the aggregate data if eachModifierelement? (default:sequenceData = FALSE)compareType: a valid score type to use for the comparison. IfsequenceData = FALSEthis defaults tomainScore(x), whereas ifsequenceData = TRUEall columns will be used by settingallTypes = TRUE.allTypes: TRUE or FALSE? Should all available score be compared? (default:allTypes = sequenceData)...: passed on tosubsetByCoord
- coord
coordinates of position to subset to. Either a
GRangesor aGRangesListobject. For both types the 'Parent' column is expected to match the transcript name. TheGRangesListobject is unlisted and only non duplicated entries are retained.- normalize
either a single logical or character value. If it is a character, it must match one of the names in the
ModifierSet.
Value
compareByCoord returns a
DataFrame and
plotCompareByCoord returns a ggplot object, which can be
modified further. The DataFrame contains columns per sample as well
as the columns names, positions and mod incorporated
from the coord input. If coord contains a column
Activity this is included in the results as well.
Examples
data(msi,package="RNAmodR")
# constructing a GRanges obejct to mark positive positions
mod <- modifications(msi)
coord <- unique(unlist(mod))
coord$score <- NULL
coord$sd <- NULL
# return a DataFrame
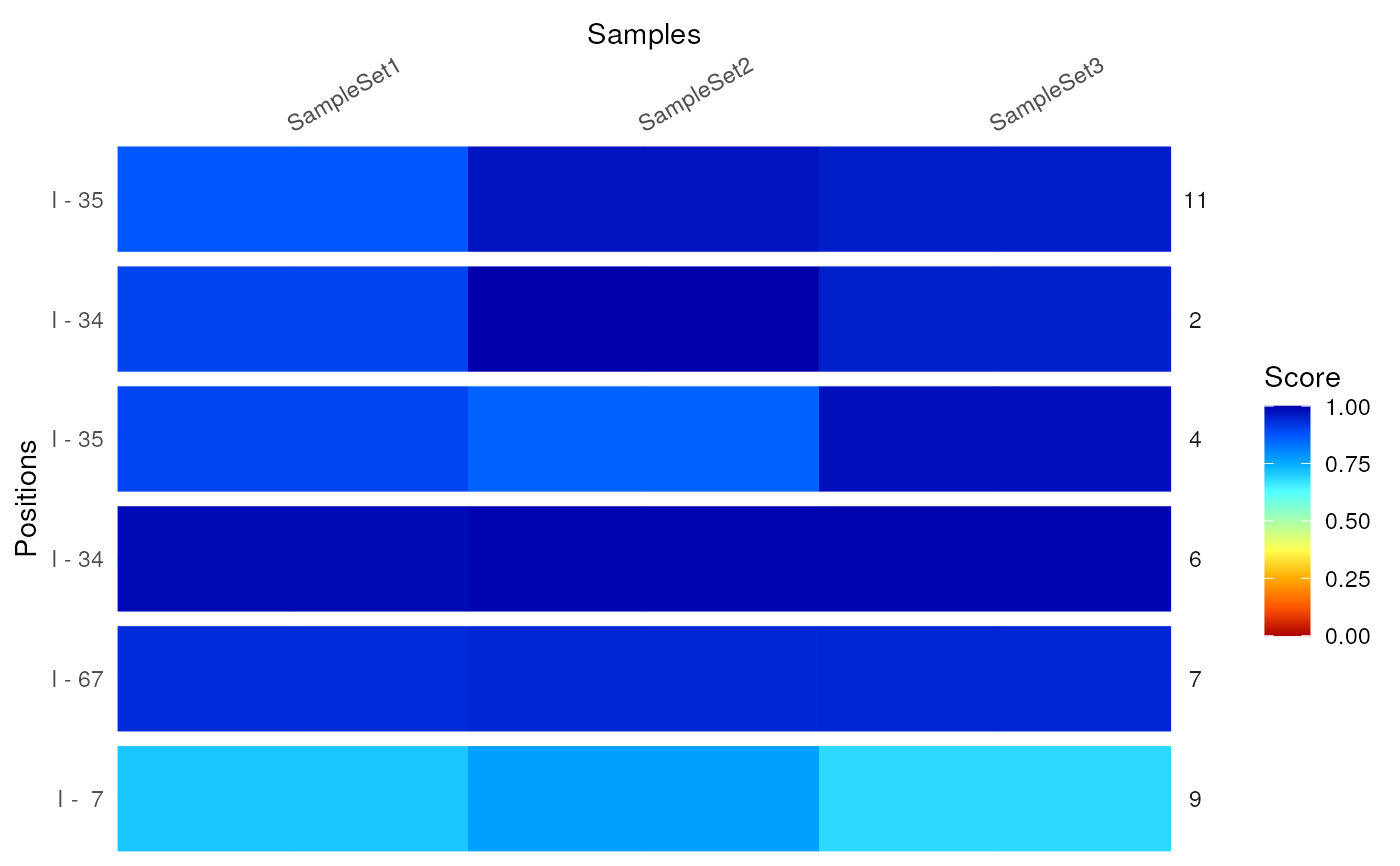
compareByCoord(msi,coord)
#> DataFrame with 6 rows and 6 columns
#> SampleSet1 SampleSet2 SampleSet3 names positions mod
#> <numeric> <numeric> <numeric> <factor> <factor> <character>
#> 1 0.900932 0.998134 0.953651 2 34 I
#> 2 0.899622 0.856241 0.976928 4 35 I
#> 3 0.984035 0.992012 0.993128 6 34 I
#> 4 0.934553 0.942905 0.943773 7 67 I
#> 5 0.709758 0.766484 0.681451 9 7 I
#> 6 0.874027 0.971474 0.954782 11 35 I
# plot the comparison as a heatmap
plotCompareByCoord(msi,coord)