Analyzing tRNA sequences and structures
Felix G.M. Ernst
2025-10-05
Source:vignettes/tRNA.Rmd
tRNA.RmdAbstract
Example of importing tRNAdb output as GRanges
Introduction
The tRNA package provides access to tRNA feature
information for subsetting and visualization. Visualization functions
are implemented to compare feature parameters of multiple tRNA sets and
to correlate them to additional data.
As input the package expects a GRanges object with
certain metadata columns. The following columns are required:
tRNA_length, tRNA_type,
tRNA_anticodon, tRNA_seq,
tRNA_str, tRNA_CCA.end. The
tRNA_str column must contain a valid dot bracket
annotation. For more details please have a look at the vignette of the
Structstrings package.
Loading tRNA information
To work with the tRNA package, tRNA information can be
retrieved or loaded into a R session in a number of ways:
- A
GRangesobject can be constructed manually containing the required colums mentioned above. - a tRNAscan result file can be loaded using the function
import.tRNAscanAsGRanges()from thetRNAscanImportpackage - selected tRNA information can be retrieved using the function
import.tRNAdb()from thetRNAdbImportpackage
For the examples in this vignette a number of predefined
GRanges objects are loaded.
library(tRNA)
library(Structstrings)
data("gr", package = "tRNA")tRNA sequences and structures
To retrieve the sequences for individual tRNA structure elements the
functions gettRNAstructureGRanges or
gettRNAstructureSeqs can be used. Several optional
arguments can be used to modify the result (See
?gettRNAstructureSeqs).
# just get the coordinates of the anticodonloop
gettRNAstructureGRanges(gr, structure = "anticodonLoop")## $anticodonLoop
## IRanges object with 299 ranges and 0 metadata columns:
## start end width
## <integer> <integer> <integer>
## TGG 31 37 7
## TGC 32 38 7
## CAA 31 37 7
## AGA 31 37 7
## TAA 31 37 7
## ... ... ... ...
## CAT 32 38 7
## GAA 31 37 7
## TTA 31 37 7
## TAC 32 38 7
## CAT 32 38 7
gettRNAstructureSeqs(gr, joinFeatures = TRUE, structure = "anticodonLoop")## $anticodonLoop
## RNAStringSet object of length 299:
## width seq names
## [1] 7 UUUGGGU TGG
## [2] 7 CUUGCAA TGC
## [3] 7 UUCAAGC CAA
## [4] 7 UUAGAAA AGA
## [5] 7 CUUAAGA TAA
## ... ... ...
## [295] 7 CUCAUAA CAT
## [296] 7 UUGAAGA GAA
## [297] 7 UUUUAGU TTA
## [298] 7 UUUACAC TAC
## [299] 7 GUCAUGA CATIn addition, the sequences can be returned already joined to get a
fully blank padded set of sequences. The boundaries of the individual
structures is returned as metadata of the RNAStringSet
object.
seqs <- gettRNAstructureSeqs(gr[1L:10L], joinCompletely = TRUE)
seqs## RNAStringSet object of length 10:
## width seq
## [1] 85 GGGCGUGUGGUC-UAGU-GGUAU-GAUUCUCGC...------GCCUGGGUUCAAUUCCCAGCUCGCCCC
## [2] 85 GGGCACAUGGCGCAGUU-GGU-AGCGCGCUUCC...------GCAUCGGUUCGAUUCCGGUUGCGUCCA
## [3] 85 GGUUGUUUGGCC-GAGC-GGUAA-GGCGCCUGA...AA-GAUGCAAGAGUUCGAAUCUCUUAGCAACCA
## [4] 85 GGCAACUUGGCC-GAGU-GGUAA-GGCGAAAGA...U-GCCCGCGCAGGUUCGAGUCCUGCAGUUGUCG
## [5] 85 GGAGGGUUGGCC-GAGU-GGUAA-GGCGGCAGA...UUGUCCGCGCGAGUUCGAACCUCGCAUCCUUCA
## [6] 85 GCGGAUUUAGCUCAGUU-GGG-AGAGCGCCAGA...------GCCUGUGUUCGAUCCACAGAAUUCGCA
## [7] 85 GGUCUCUUGGCC-CAGUUGGUAA-GGCACCGUG...------ACAGCGGUUCGAUCCCGCUAGAGACCA
## [8] 85 GCGCAAGUGGUUUAGU--GGU-AAAAUCCAACG...-------CCCCGGUUCGAUUCCGGGCUUGCGCA
## [9] 85 GGCAACUUGGCC-GAGU-GGUAA-GGCGAAAGA...U-GCCCGCGCAGGUUCGAGUCCUGCAGUUGUCG
## [10] 85 GCUUCUAUGGCC-AAGUUGGUAA-GGCGCCACA...------ACAUCGGUUCAAAUCCGAUUGGAAGCA
# getting the tRNA structure boundaries
metadata(seqs)[["tRNA_structures"]]## IRanges object with 15 ranges and 0 metadata columns:
## start end width
## <integer> <integer> <integer>
## acceptorStem.prime5 1 7 7
## Dprime5 8 9 2
## DStem.prime5 10 13 4
## Dloop 14 23 10
## DStem.prime3 24 27 4
## ... ... ... ...
## TStem.prime5 61 65 5
## Tloop 66 72 7
## TStem.prime3 73 77 5
## acceptorStem.prime3 78 84 7
## discriminator 85 85 1Be aware, that gettRNAstructureGRanges and
gettRNAstructureSeqs might not be working as expected, if
the tRNA sequences in questions are armless or deviate drastically from
the canonical tRNA model. The functions in the tRNA
packages were thouroughly tested using human mitochondrial tRNA and
other tRNAs missing certain features. However, for fringe cases results
may differ. If you encounter such a case, please report it with an
example.
Subsetting tRNA sequences
Structure information of the tRNA can be queried for subsetting using
several functions. For the following examples the functions
hasAccpeptorStem and hasDloop are used.
gr[hasAcceptorStem(gr, unpaired = TRUE)]
# mismatches and bulged are subsets of unpaired
gr[hasAcceptorStem(gr, mismatches = TRUE)]
gr[hasAcceptorStem(gr, bulged = TRUE)]
# combination of different structure parameters
gr[hasAcceptorStem(gr, mismatches = TRUE) &
hasDloop(gr, length = 8L)]Please have a look at the man page ?hasAccpeptorStem for
all available subsetting functions.
Visualization
To get an overview of tRNA features and compare different datasets,
the function gettRNAFeaturePlots is used. It accepts a
named GRangesList as input. Internally it will calculate a
list of features values based on the functions mentioned above and the
data contained in the mcols of the GRanges objects.
# load tRNA data for E. coli and H. sapiens
data("gr_eco", package = "tRNA")
data("gr_human", package = "tRNA")
# get summary plots
grl <- GRangesList(Sce = gr,
Hsa = gr_human,
Eco = gr_eco)
plots <- gettRNAFeaturePlots(grl)## Warning: `aes_()` was deprecated in ggplot2 3.0.0.
## ℹ Please use tidy evaluation idioms with `aes()`
## ℹ The deprecated feature was likely used in the tRNA package.
## Please report the issue at <https://github.com/FelixErnst/tRNA/issues>.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.
plots$length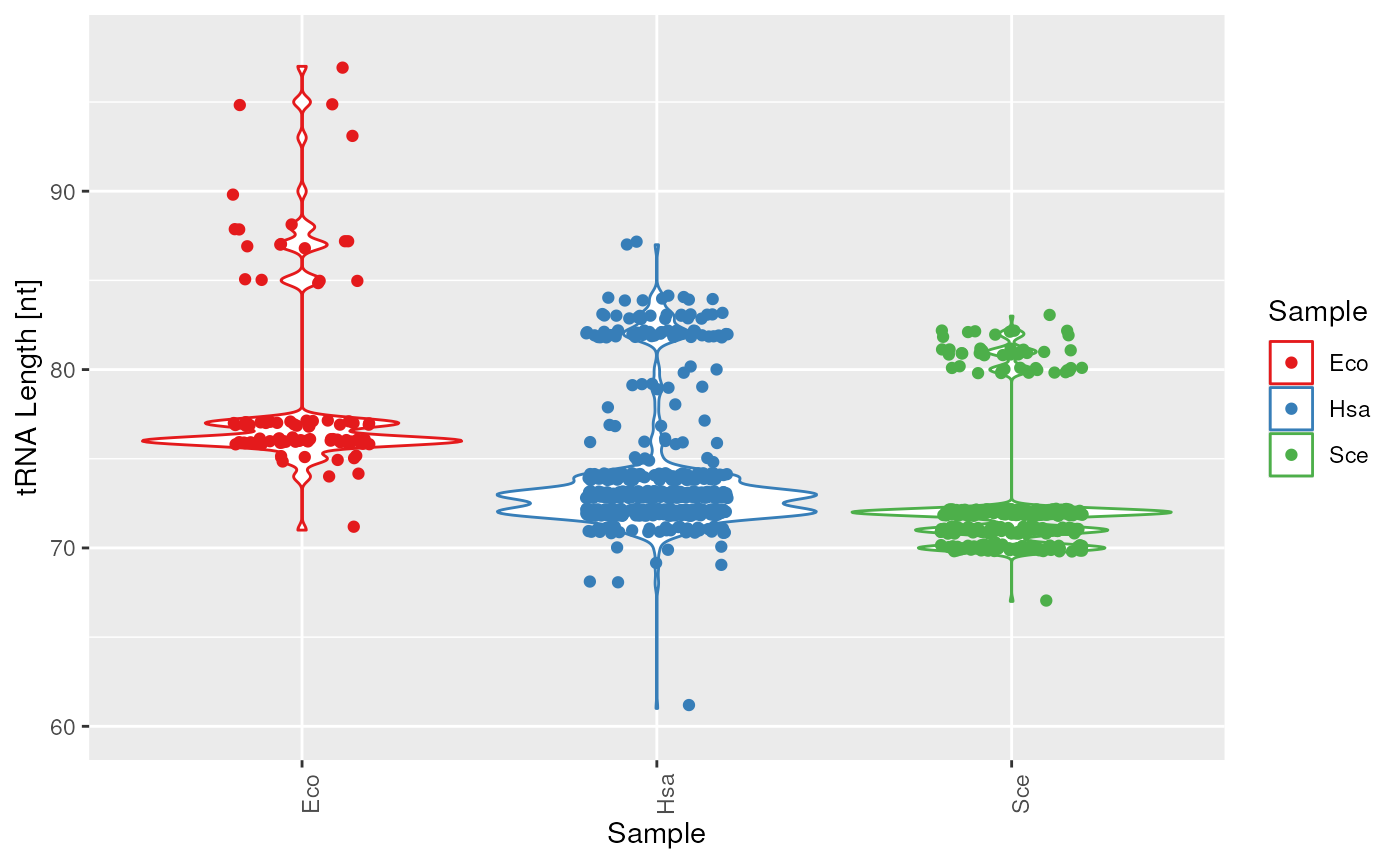
tRNA length.
plots$tRNAscan_score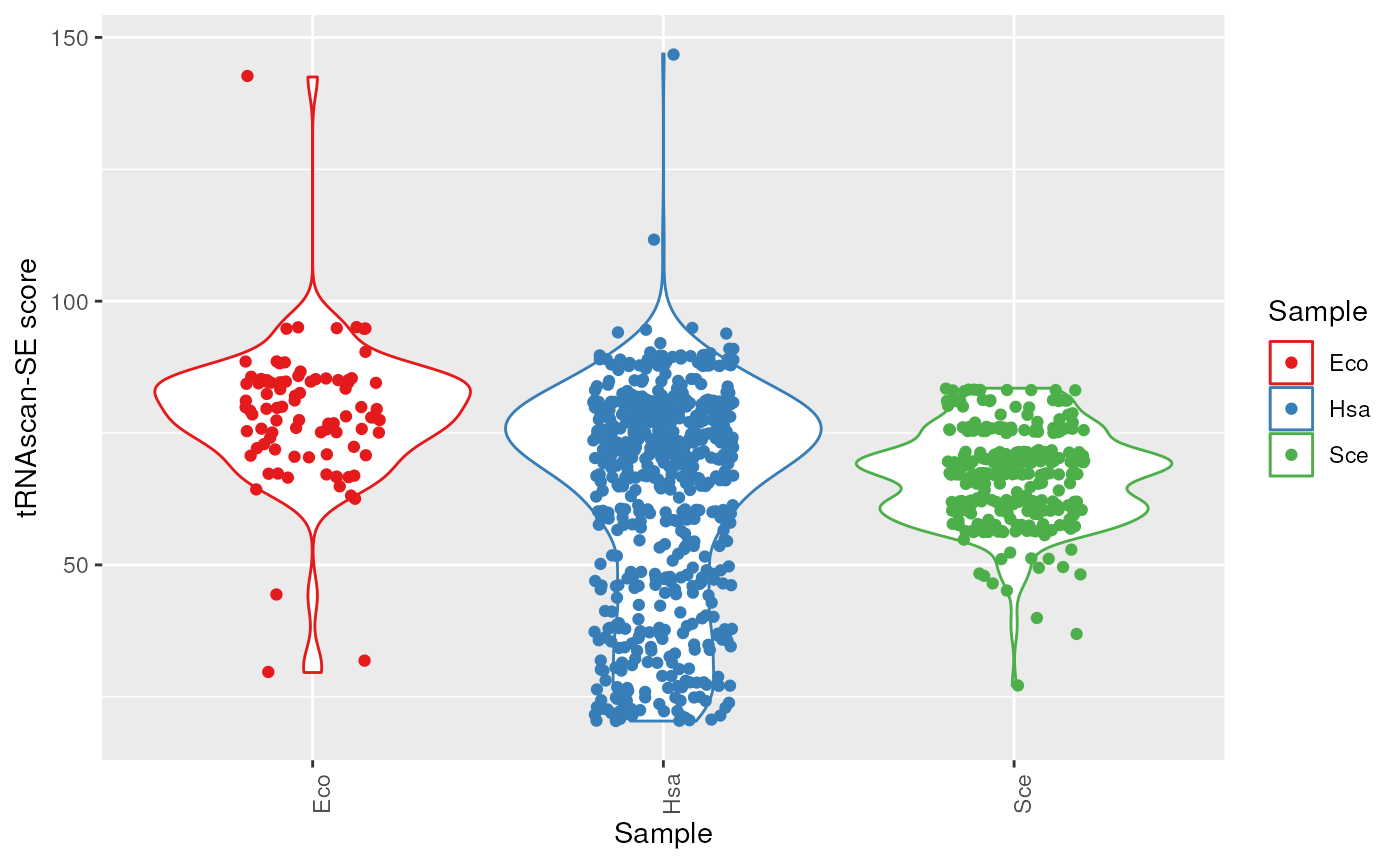
tRNAscan-SE scores.
plots$gc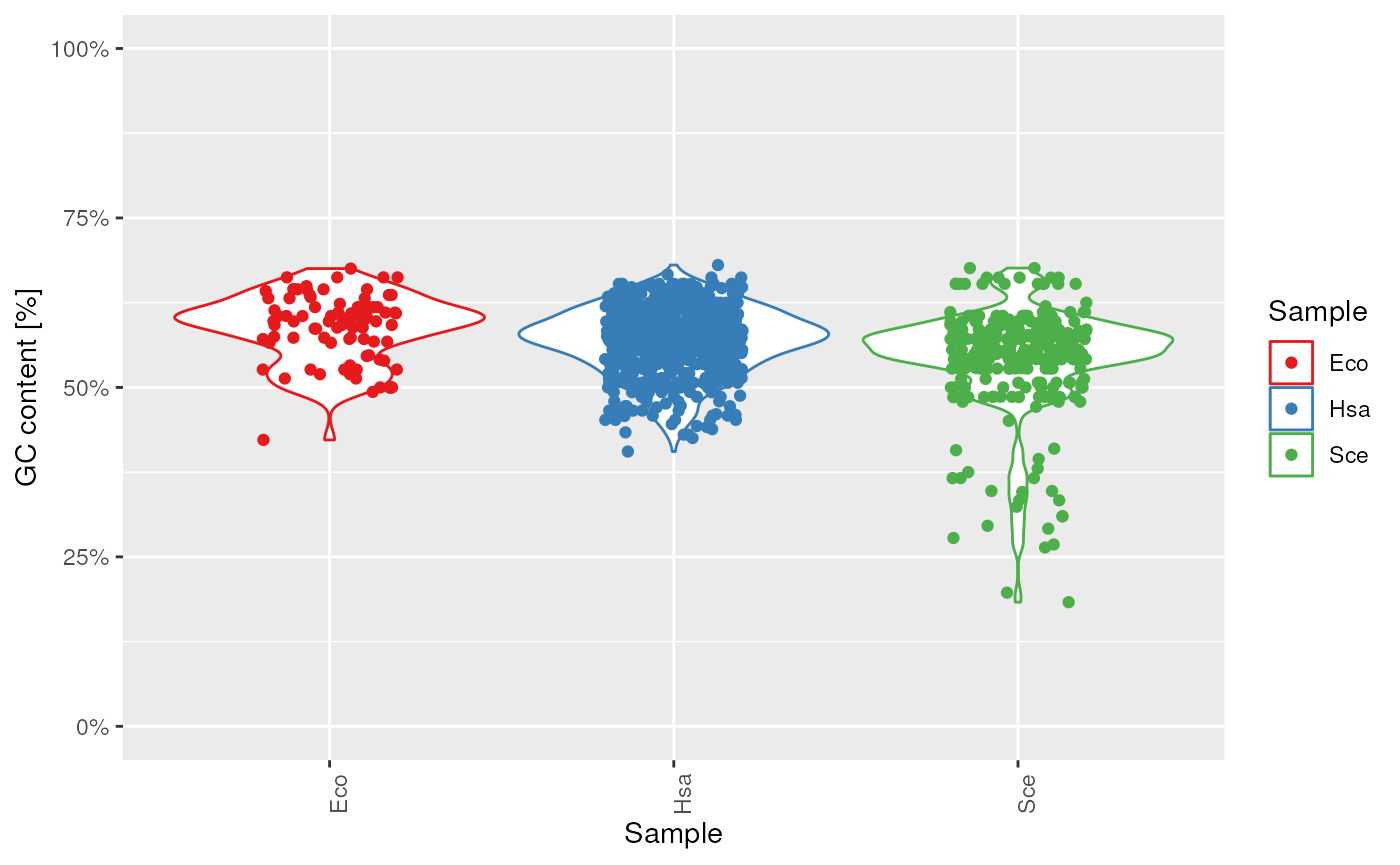
tRNA GC content.
plots$tRNAscan_intron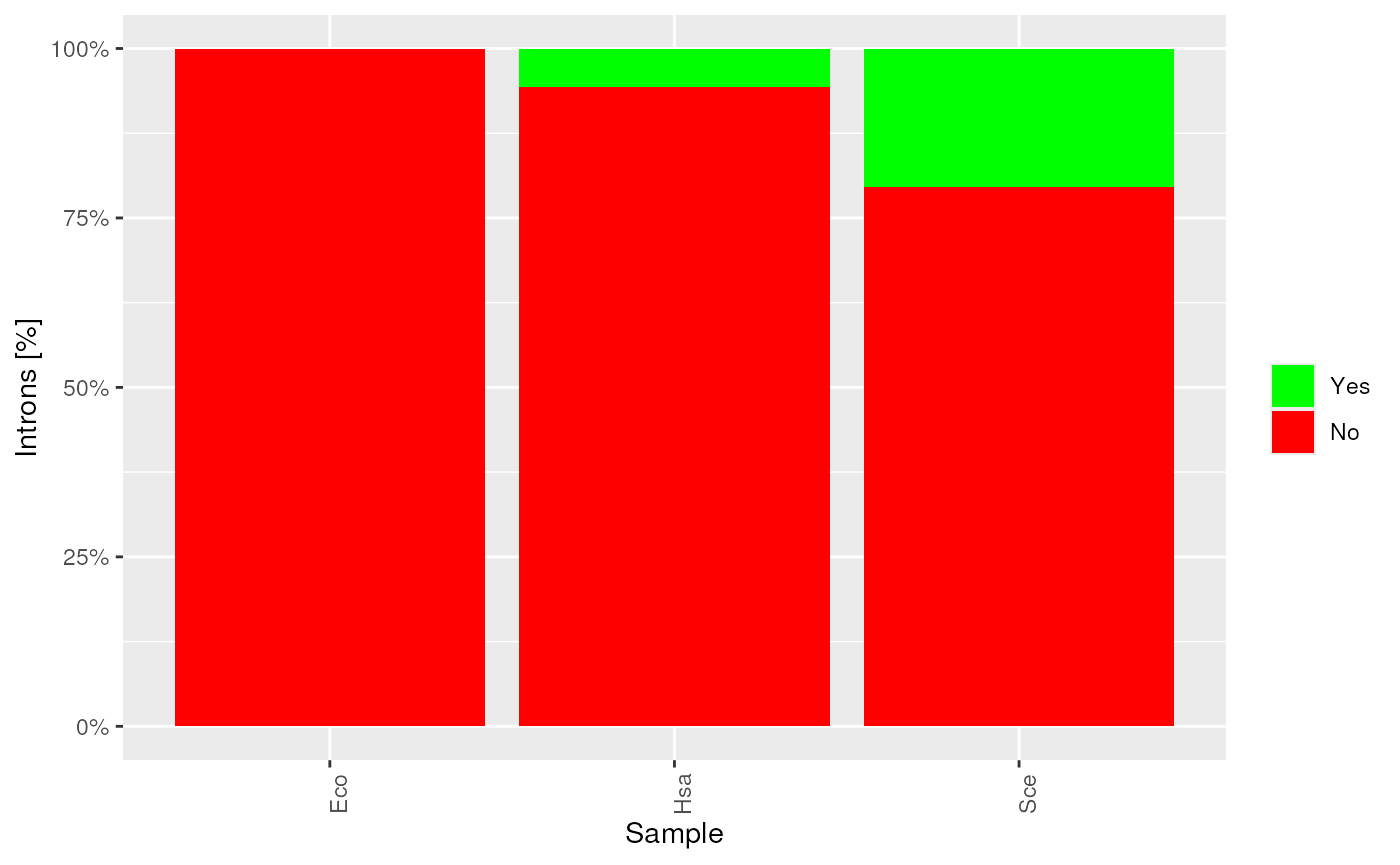
tRNAs with introns.
plots$variableLoop_length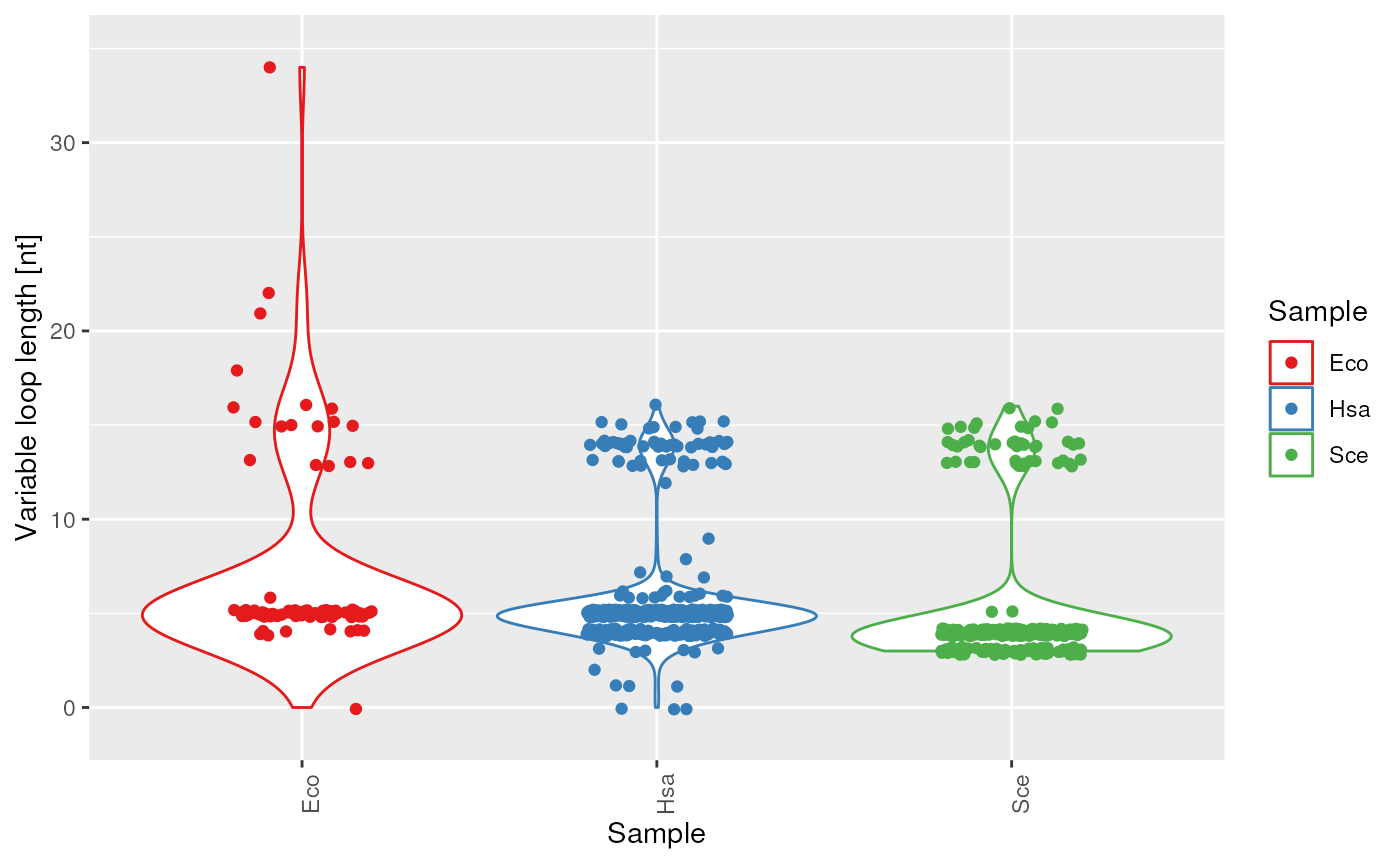
Length of the variable loop.
To access the results without generating plots, use the function
gettRNASummary.
To check whether features correlate with additional scores, optional
arguments can be added to gettRNAFeaturePlots or used from
the score column of the GRanges objects. For
the first case a list of scores with the same dimensions as the
GRangesList object has to be provided as the argument
scores. For the latter case, just set the argument
plotScore = TRUE.
# score column will be used
plots <- gettRNAFeaturePlots(grl, plotScores = TRUE)
plots <- gettRNAFeaturePlots(grl,
scores = list(runif(length(grl[[1L]]),0L,100L),
runif(length(grl[[2L]]),0L,100L),
runif(length(grl[[3L]]),0L,100L)))
plots$length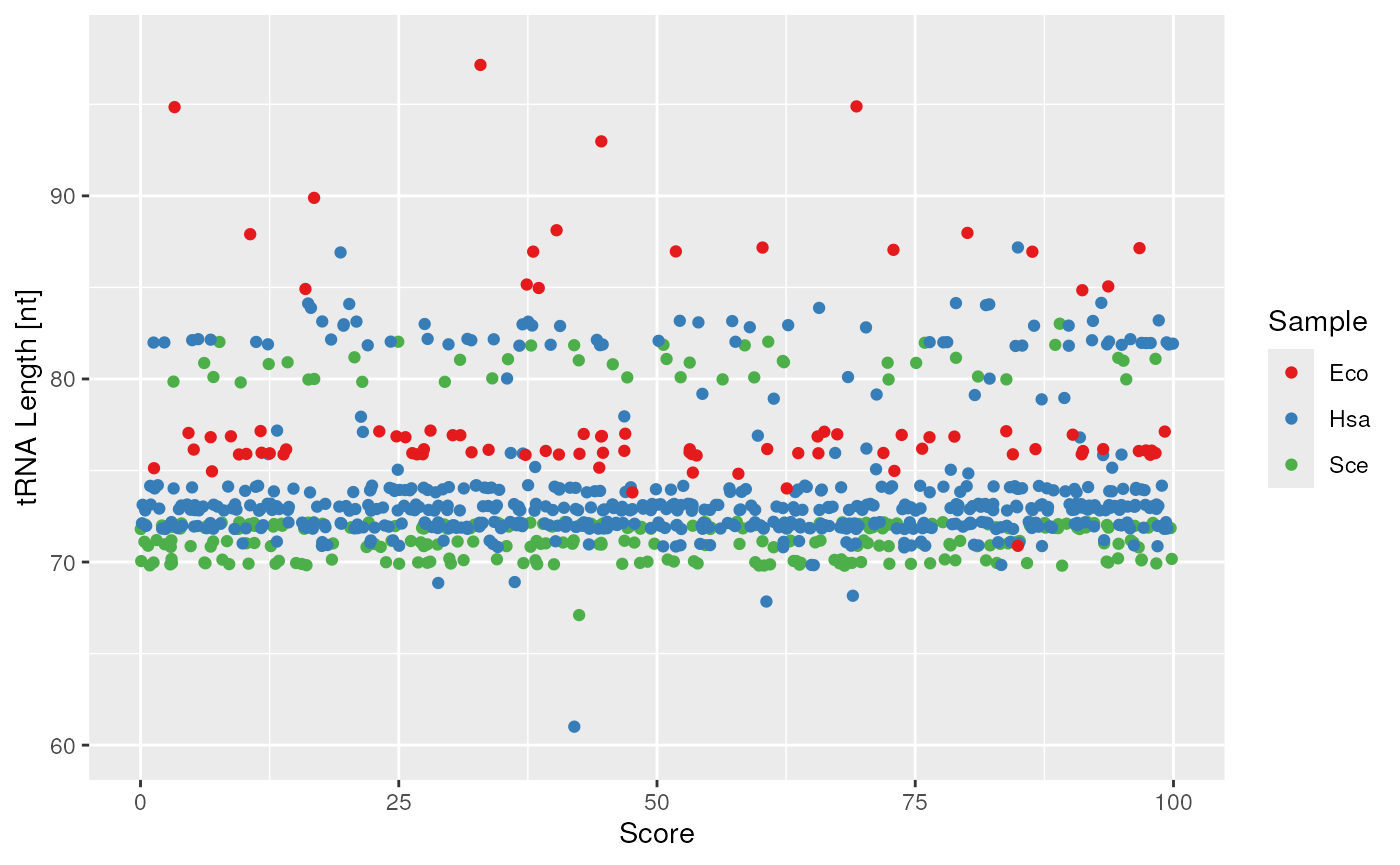
tRNA length and score correlation.
plots$variableLoop_length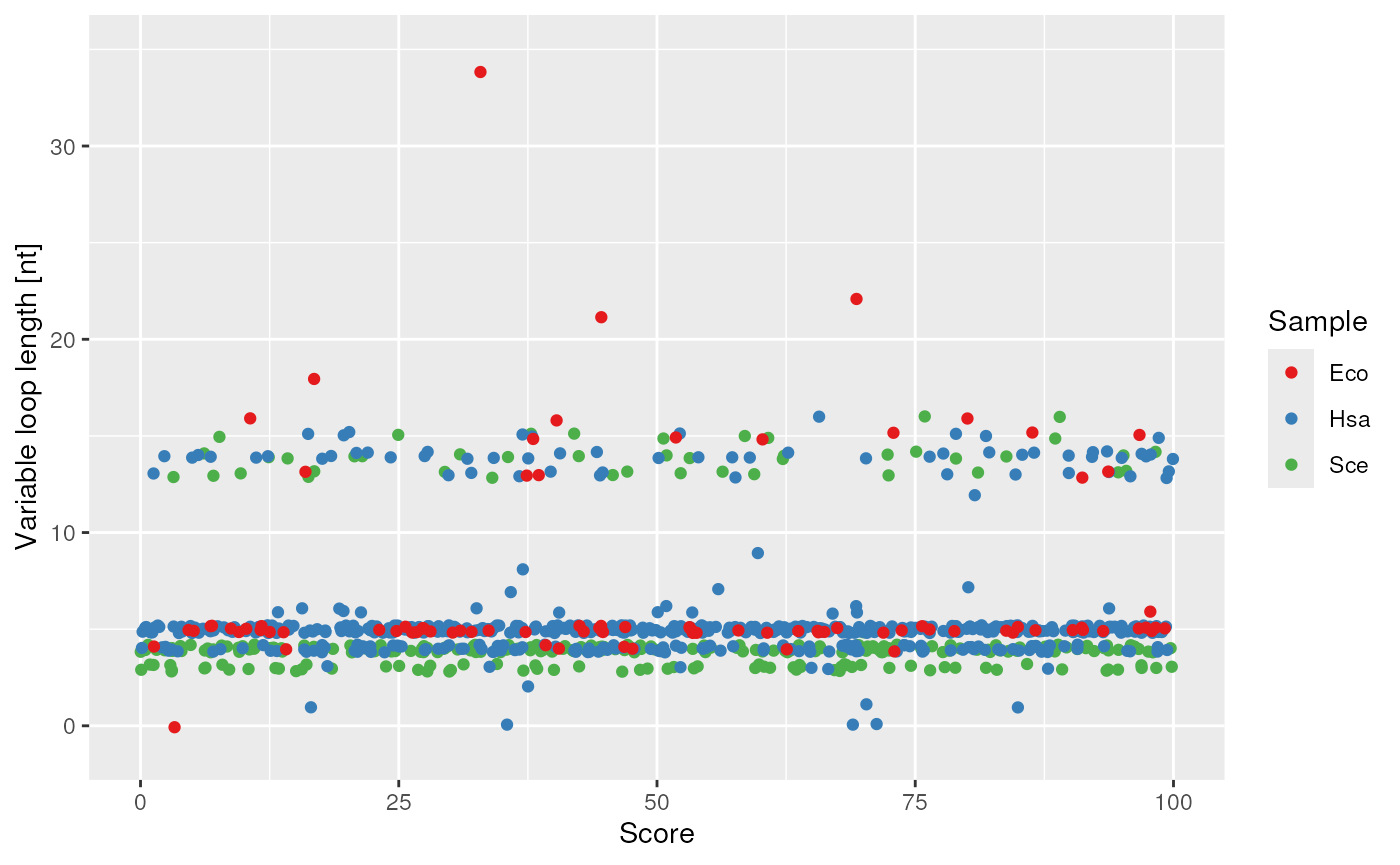
variable loop length and score correlation.
Since all plots returned by the functions mentioned above are
ggplot2 objects, they can be modified manually and changed
to suit your needs.
plots$length$layers <- plots$length$layers[c(-1L,-2L)]
plots$length + ggplot2::geom_boxplot()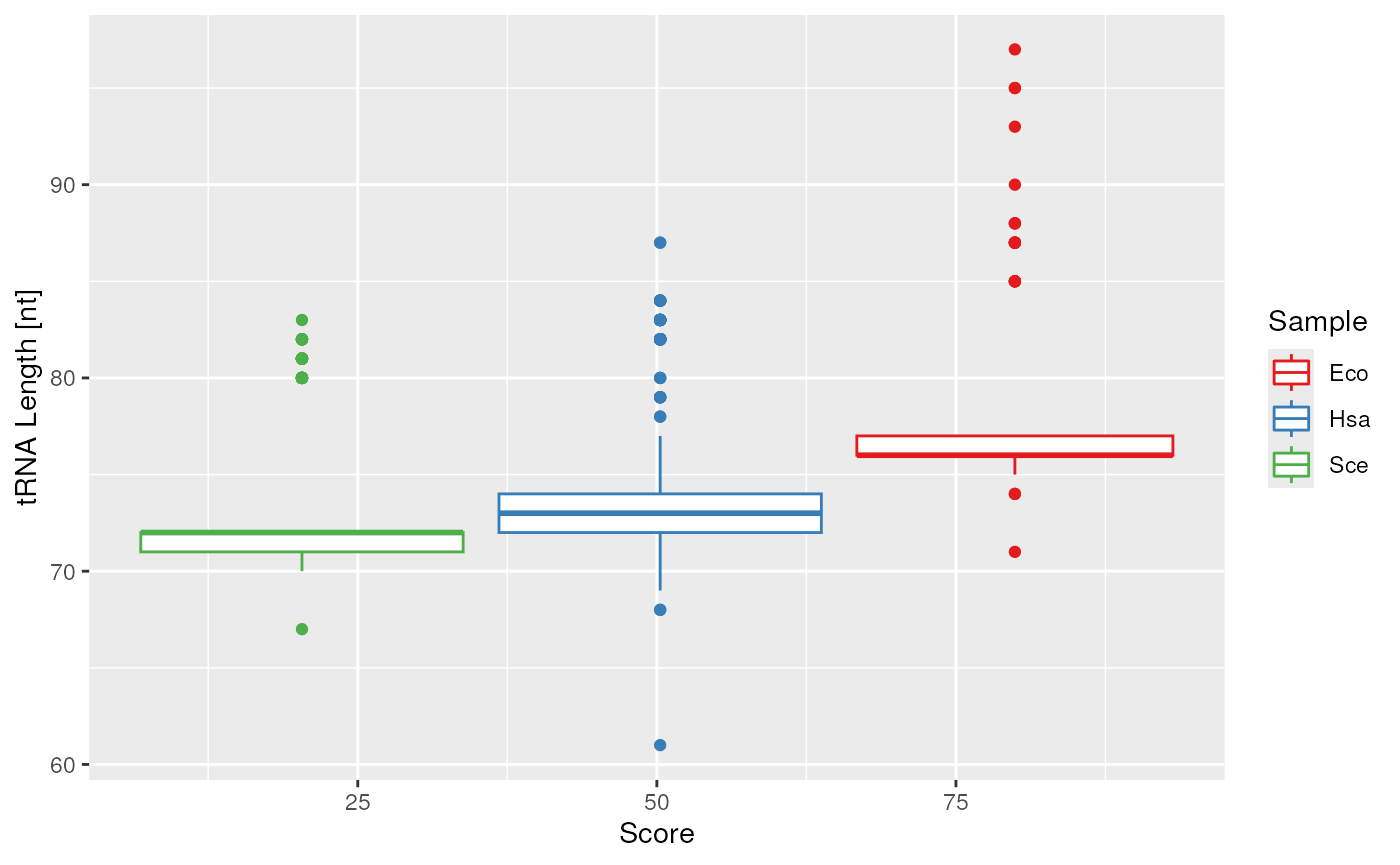
Customized plot switching out the point and violin plot into a boxplot.
In addition, the data of the plots can be accessed directly.
head(plots$length$data)## id value score
## 1 Sce 71 73.288199
## 2 Sce 72 77.252151
## 3 Sce 80 87.460066
## 4 Sce 81 17.494063
## 5 Sce 82 3.424133
## 6 Sce 72 32.038573Options
The colours of the plots can be customized directly on creation with the following options.
options("tRNA_colour_palette")## $tRNA_colour_palette
## [1] "Set1"
options("tRNA_colour_yes")## $tRNA_colour_yes
## [1] "green"
options("tRNA_colour_no")## $tRNA_colour_no
## [1] "red"Dot bracket annotation
To retrieve detailed information on the base pairing the function
gettRNABasePairing() is used. Internally this will
construct a DotBracketStringSet from the
tRNA_str column, if this column does not already contain a
DotBracketStringSet. It is then passed on to the
Structstrings::getBasePairing function.
A valid DotBracket annotation is expected to contain only pairs of
<>{}[]() and the . character (Please
note the orientation. For <> the orientation is
variable, since the tRNAscan files use the ><
annotation by default. However upon creation of a
DotBracketStringSet this annotation will be converted).
head(gettRNABasePairing(gr)[[1L]])## DotBracketDataFrame with 6 rows and 4 columns
## pos forward reverse character
## <integer> <integer> <integer> <character>
## 1 1 1 70 <
## 2 2 2 69 <
## 3 3 3 68 <
## 4 4 4 67 <
## 5 5 5 66 <
## 6 6 0 0 .
head(getBasePairing(gr[1L]$tRNA_str)[[1L]])## DotBracketDataFrame with 6 rows and 4 columns
## pos forward reverse character
## <integer> <integer> <integer> <character>
## 1 1 1 70 <
## 2 2 2 69 <
## 3 3 3 68 <
## 4 4 4 67 <
## 5 5 5 66 <
## 6 6 0 0 .The loop ids for the structure elements can be retrieved with the
gettRNALoopIDs() function, which relies on the
Structstrings::getLoopIndices function. (For more details,
please have a look at the ?getLoopIndices)
gettRNALoopIDs(gr)[[1L]]## [1] 1 2 3 4 5 5 6 6 6 7 8 9 9 9 9 9 9 9 9 9 9 9 8 7 6
## [26] 10 11 12 13 14 14 14 14 14 14 14 14 14 13 12 11 10 6 6 6 6 15 16 17 18
## [51] 19 19 19 19 19 19 19 19 19 18 17 16 15 6 5 5 4 3 2 1 0
getLoopIndices(gr[1L]$tRNA_str)## LoopIndexList of length 1
## [[""]] 1 2 3 4 5 5 6 6 6 7 8 9 9 9 9 9 ... 19 19 19 18 17 16 15 6 5 5 4 3 2 1 0Session info
## R version 4.5.1 (2025-06-13)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] tRNA_1.27.1 Structstrings_1.25.3 Biostrings_2.77.2
## [4] XVector_0.49.1 GenomicRanges_1.61.5 Seqinfo_0.99.2
## [7] IRanges_2.43.5 S4Vectors_0.47.4 BiocGenerics_0.55.1
## [10] generics_0.1.4 BiocStyle_2.37.1
##
## loaded via a namespace (and not attached):
## [1] sass_0.4.10 stringi_1.8.7 digest_0.6.37
## [4] magrittr_2.0.4 evaluate_1.0.5 grid_4.5.1
## [7] RColorBrewer_1.1-3 bookdown_0.45 fastmap_1.2.0
## [10] jsonlite_2.0.0 BiocManager_1.30.26 scales_1.4.0
## [13] textshaping_1.0.3 jquerylib_0.1.4 cli_3.6.5
## [16] rlang_1.1.6 crayon_1.5.3 withr_3.0.2
## [19] cachem_1.1.0 yaml_2.3.10 tools_4.5.1
## [22] dplyr_1.1.4 ggplot2_4.0.0 vctrs_0.6.5
## [25] R6_2.6.1 lifecycle_1.0.4 stringr_1.5.2
## [28] fs_1.6.6 Modstrings_1.25.0 htmlwidgets_1.6.4
## [31] ragg_1.5.0 pkgconfig_2.0.3 desc_1.4.3
## [34] pkgdown_2.1.3 bslib_0.9.0 pillar_1.11.1
## [37] gtable_0.3.6 glue_1.8.0 systemfonts_1.3.1
## [40] tidyselect_1.2.1 xfun_0.53 tibble_3.3.0
## [43] knitr_1.50 farver_2.1.2 htmltools_0.5.8.1
## [46] labeling_0.4.3 rmarkdown_2.30 compiler_4.5.1
## [49] S7_0.2.0